Exome Sequence Assembly Utilizing Bowtie & Samtools

At the end of all the wet chemistry for a genome sequencing project we are left with the raw data in the form of fastq files. The following post documents the processing of said raw files to assembled genomes using Bowtie & Samtools.

Fig.1: Raw data is split into approximately 20-30 fastq files per individual. #
Each of these raw files, once uncompressed, contains somewhere around 1 gigabyte of nucleotide, machine, and quality information. Which will follow the fastq guidelines and look very similar to the following. It’s quickly noticeable where our nucleotide data consisting of ATGC lives within these raw files.
@HWI-ST1027:182:D1H4LACXX:5:2306:21024:142455 1:N:0:ACATTGGATTTGAATGGCACTGAATATACAGATCAACTTGAAGATAACTGATATCTAAACTATGCTGAGTCTTCTAATTCATGAACACAGTACATTTCTATTTAGG
+
@?<DFEDEHHFHDHEEGGECHHIIIIIGIGIIFGIBGHGBHGIE9>GIIIIIIIIIIIFGEII@DCHIIIIIIGHHIIFEGHBHECHEHFEDFDFDCEE>
@HWI-ST1027:182:D1H4LACXX:5:2306:21190:142462 1:N:0:ACATTGGCCCTTTTCTCTCCCAGGTGGGAGGCAGATAGCCTTGGGCAAATTTTCAAGCCCATCTCGCACTCTGCCTGGAAACAGACTCAGGGCTATTGTGGCGGGG
+
CCCFFFFFHHHHHJJJJJEGIJHIJJJIJHIJJJJJJJJJJIJJJJIJJJJIJJJJJJIIJHHHFFFFFFEDEEECCDDDDDDDDDDDDDDDEDDBDDB#
At this point the raw reads need to be assembled into contiguous overlapping sets, then chromosomes, and finally the entire genome. There are two general approaches here, template-based and de novo assembly. For this particular exome data set it is prudent to move forward with template-based assembly using the latest build of the human reference genome. An index of the reference genome must be built for bowtie, some indexes are also available for download though the file size can be quite large.
$ bowtie-build /Users/mokas/Desktop/codebase/max/hg19.fa hg19
Settings:
Line rate: 6 (line is 64 bytes)
Lines per side: 1 (side is 64 bytes)
Offset rate: 5 (one in 32)
FTable chars: 10
...
Getting block 6 of 6
Reserving size (543245712) for bucket
Calculating Z arrays
Calculating Z arrays time: 00:00:00
Entering block accumulator loop:
10%
20%
...
numSidePairs: 6467211
numSides: 12934422
numLines: 12934422
Total time for backward call to driver() for mirror index: 02:00:28
The entire reference build should be complete within an hour or two, which may be faster than downloading an pre-built index. At this point the raw fastq file is ready to be processed using our indexed template.
$ bowtie -S [ref] [fastq] [output.sam]
At the end of this step we will have a .sam (Sequence Alignment Map) file, which will have each of our raw reads aligned to certain positions on the human reference. However, the reads will be in no useful order, and all the chromosomes and locations are mixed together.

Fig.2 #
To be able to move through such a large file with speed and ease it must be converted into a binary format, at which point all the reads can be sorted into a meaningful manner.
$ samtools view -bS -o [output.bam] [input.sam]
$ samtools sort [input.bam] [output.sorted]
We are now left with a useful file where our raw reads are assembled and sorted based on a template.
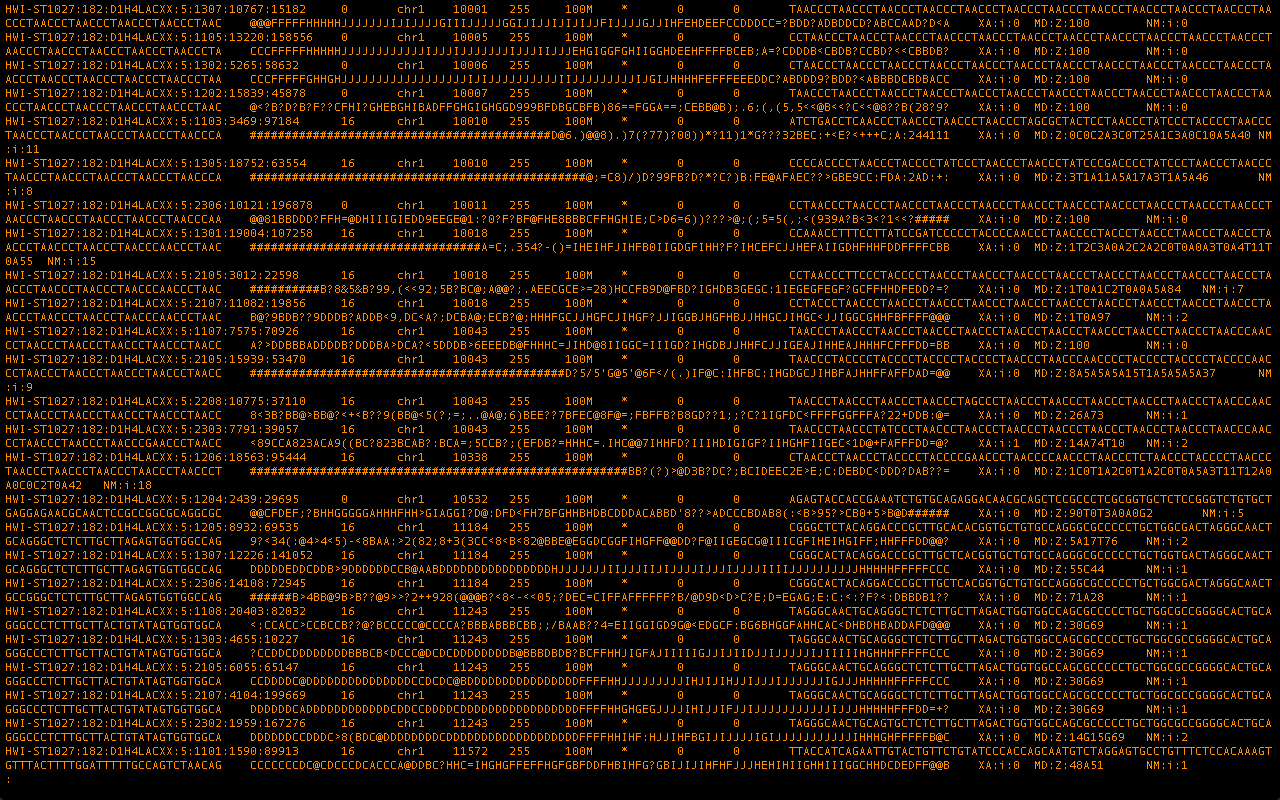
Fig.3 #
This file can be visualized and analyzed in a wide variety of available programs, the format is also accessible enough to quickly build your own tools around it. Once each of the 20-30 fastq files in a single sample have been processed in this manner the files can be merged, converted into binary for reduced file size, and indexed for quick browsing.
